中国科学技术大学生命科学学院
刘海燕 陈泉 龙朋朋 黄斌 许洋 陈耀晞
合成生物学研究用现代工程学方式构建人工生物系统,以更全面地发现和验证生物系统的设计原理,并服务社会需求。如同天然生物系统,在合成生物系统中,蛋白质仍然是最主要的功能执行者。例如,在感知处理环境信息并做出适应性响应的智能细胞中,负责信号感知的受体和负责整合内外环境信号、决定激活或抑制特定细胞功能的信号传导蛋白和转录因子等,绝大部分都是蛋白质;在用于人工生物合成的细胞工厂中,代谢通路中的生化反应都需要作为酶的蛋白质来催化。
目前,绝大多数人工生物系统是基于天然蛋白质元件及其已知相互作用方式来构建的。例如,原核体系中,不同功能基因线路广泛使用少数几种已被较好表征的转录因子作为基因开关;人工生物合成路线主要用天然酶完成对特定底物的特定催化步骤等。这种方法的优点是,基于对天然蛋白质性质相对较为深入的了解,研究工作可能有较高的效率和成功率。然而,如果仅限于利用已知的天然蛋白质元件,合成生物体系的规模、适用范围等会受到极大限制,如体系能响应的化学信号有限;人工生物合成的底物选择性、反应选择性等受制于可用的酶;此外,环境对人工体系的干扰或人工体系不同部分之间的干扰受制于天然蛋白质的特性,且随系统规模增大,这种干扰的可能性快速增加。
克服上述困难有两条可能的途径。第一条途径是,从自然界中存在的大量具有不同功能,特别是具有不同特异性分子识别能力的天然蛋白质中找出适当的元件。例如,同一家族的不同转录因子可响应不同的化学信号,也可识别不同的操纵子序列;催化同类化学反应的酶可能有不同的底物专一性和反应特异性,等等。另一条途径是,采用蛋白质设计和定向进化等手段,改变天然蛋白质分子的功能活性,甚至重新设计蛋白质,获得适用的元件。例如,通过诱导物结合位点或 DNA 结合位点改造,转录因子可以响应新的化学诱导信号或识别新的 DNA 结合位点;酶的底物特异性可通过蛋白质工程改变等。
上述两条途径分别对应蛋白质元件发现和元件设计改造。由于已知的天然蛋白质元件难以满足合成生物学对元件功能活性的多样性需求,蛋白质元件发现和改造是合成生物学必不可少的研究内容。目前,此方向的大多数研究还是以实验手段为主。例如,在元件发现中,通过实验筛选鉴定转录因子识别的诱导物和操纵子、酶催化的底物;在元件改造中,通过定向进化改变酶的底物特异性,甚至催化反应类型,以及通过蛋白质结构域融合引入新的调控方式等。纯粹的实验手段存在时间成本高、耗费资源多、通量有限、从无到有获得新功能极为困难等不利因素。计算和数据驱动的方法是克服这些不利因素的重要途径。随着计算生物学的发展以及生物学数据的积累,实验和计算的有机结合将会是蛋白质元件发现和改造的最有效途径。
在本文中,我们将讨论蛋白质元件发现和改造中计算的必要性,举例说明研究方法和工具,在此过程中探讨现有方法的主要困难和局限,以及可能的解决途径等。蛋白质元件发现和设计需要生物信息学、化学信息学、分子模拟等不同方面计算工具的综合应用和创新。考虑到对这些不同方面计算方法的进展综述已经比较多,且本文篇幅有限,我们将不试图综述和总结不同方法工具的进展,而仅仅以转录因子元件发现、催化元件发现和设计为例,较为概括性地展示计算方法在蛋白质元件发现和设计中可以发挥怎样的作用,并尝试前瞻性地讨论计算应聚焦的重点问题。希望通过本文的讨论,能更好地促进实验和计算的有机结合,推动合成生物学中的蛋白质元件预测和设计研究。
1 计算在蛋白质元件发现和设计中的必要性
基于基因测序数据,我们已知道了大量天然蛋白质的氨基酸序列;随着蛋白质空间结构数据的积累,对多数未知结构天然蛋白质也有可能找到其同源蛋白的空间结构。然而,我们对高度多样化的潜在天然蛋白质元件的了解大多停留于此水平。在为特定合成生物系统选择蛋白质元件时,仅仅基于蛋白质自身的序列和结构信息难以判断一种天然蛋白质是否是合适的元件。
为更具体地说明这一问题,我们以原核生物中的 tetR (四环素阻遏蛋白 )家族转录因子为例。该家族蛋白在原核生物中广泛分布,家族成员参与了抗生素耐受、生物合成代谢、应激响应等不同过程的调控。在公共蛋白质数据库中已存在超过 20 万条 tetR 家族蛋白质序列信息。理论上,大量的 tetR 家族天然蛋白均可作为合成生物学的候选蛋白质元件,从而为构建有价值的人工生物系统提供高度丰富的可能性。例如,不同的 tetR蛋白能用来感应不同的化学小分子,还可以设计相互正交的 DNA调控位点等。然而,纯粹从实验获得的,关于这些蛋白质成员的信息远不能支持这样的应用:在蛋白质水平上被初步表征过的 tetR 蛋白还不到 200 种;仅不到 100 种有其化学诱导物的信息;测定空间结构的更少。由于不知道绝大部分 tetR 家族蛋白识别或可能识别什么样的小分子诱导物,也不知道它们的 DNA 结合特异性,因此就难以在合成生物体系中使用它们。靠实验分析来补全这些信息,通量将十分有限。如果计算分析能做出有价值的预测,包括对部分家族成员蛋白做出有一定可靠性的预测,可极大有利于它们在合成生物学中的应用。
另一个例子是获得代谢通路所需的酶元件。这方面要解决的一类问题是如何从大量已知其催化的反应的天然酶中自动检索出需要的酶。在这类问题中计算无疑是重要的,但由于篇幅所限,本文的讨论不包括这方面内容。另一类问题是,当从目前已知的天然酶中还找不到催化特定目标反应的酶时,如何从反应性能未知的酶中发现有可能催化目标反应的酶。计算生物学如果能基于序列和结构等信息预测天然酶催化的反应类型和底物特异性,无论是指认性预测还是排除性预测,都可能极大降低用实验方法筛选酶元件的工作量,提高成功率。
除发现有所需功能的蛋白质元件外,蛋白质元件的改造,乃至重新设计也可以极大受益于计算。对天然蛋白质进行改造的目标包括调控稳定性、改变环境偏好性、改变相互作用特异性 (如转录因子识别新的诱导物分子或 DNA 序列、酶催化新的底物等 )等;在一些应用中,可能还必须获得自然界不存在的全新功能的蛋白质,如催化新反应类型的酶。从发展的角度看,解决后一类问题是合成生物学走出天然体系的局限,达到“超越自然”目标的重要途径。
目前,实验室定向进化仍然是蛋白质元件改造的主要手段,但其限制因素包括投入高、风险大 (成功率低 )、高度依赖于问题和研究人员的经验等。此外,从无任何初始活性的蛋白质出发经实验室定向进化获得新催化反应类型、新催化机制等尚很困难,极少数成功的例子难以推广。
计算设计在蛋白质元件改造中的价值已有很多例证。在大量研究实例中,通过计算筛选突变位点和突变范围,定向进化的成功率得到极大提高;此外,通过计算从头设计酶,也已有成功实例。尽管初始设计的酶只有最低可探测的催化活性,但它们已可以作为定向进化的出发点,最终得到催化效率达天然酶水平的人工酶。随计算方法改进,理性设计将达到更高成功率,进一步降低实验需求和相应的资源耗费,并能解决更具挑战性的元件设计问题。
2 研究方法和工具举例
2.1 研究方法的综合性
用计算方法研究蛋白质元件必然涉及生物信息学、化学信息学、计算化学、结构生物学、大数据等不同领域、不同类型计算工具的综合应用。这是由问题本身的特点所决定的:因为计算能否给出有价值的结果,取决于能否最大限度地利用基因组序列及其分类和进化等关系 (生物信息学 )、化学小分子 (化学信息学 )与蛋白质的相互作用及其与蛋白质结构和动力学关系 (计算化学、结构生物学 )、从大量序列结构和相互作用数据中总结提炼的规律 (大数据 )等。目前,绝大多数计算工具在通用性、准确性等方面还做不到面面俱到,这些工具需要和相关生物学背景知识、具体应用场景结合起来,才可能在解决特定元件发现或设计问题时最大程度发挥效用。在本节其余部分,我们将分别以对 tetR 家族蛋白 DNA 识别特异性的预测和蛋白质与小分子结合口袋的设计为例,说明综合应用不同方法的原理和可能达到的效果,并从研究方法所依据的生物学假设、不同方法在通用性和准确性之间的平衡、发展新计算方法的需求等角度,探讨计算方法研究和应用中应关注的不同方面。
2.2 预测转录因子识别的 DNA 序列
首先,我们考察一类基于基因组序列预测转录因子与 DNA 结合序列的方法。该方法基于以下现象或假设:被 tetR 家族蛋白识别的 DNA 序列具有回文特征;蛋白质识别的 DNA 序列在基因组上频繁出现在相应蛋白质基因位点附近;蛋白质 DNA 结合结构域的氨基酸序列决定识别位点半回文区的 DNA 序列。基于这些假设,可相对容易地预测一些 tetR 成员的 DNA 识别位点。我们把这个过程流程化、程序化,建立计算工具,从而能对大量 tetR 家族成员做出自动预测(龙朋朋等,待发表)。这里,简单把手工流程翻译成程序不足以实现高鲁棒性的自动计算。流程自动化过程中需要考虑的问题包括:每个转录因子基因位点附近都可以找到大量的回文序列片段,但其中绝大部分 (或全部 )都不是我们要寻找的位点;要确定真正的结合位点,必须考虑其他含同源蛋白的基因组中回文序列片段在目标区域被富集的情况,在此过程中,我们需要排除目标蛋白编码区以外基因组同源性的影响;此外,回文序列的判别会受到序列碱基组成的影响,如 G、C 含量高的片段容易被识别为回文序列,且在基因组中出现频率高,等等。综合处理好这些因素后,程序化的流程能产生可重复、可靠的结果,而无需依赖手工筛选 (手工筛选难以做到高通量预测 )。图 1显示了把这一自动化流程应用于基因组序列已知,可从公共蛋白数据库中找到的全部 tetR 家族成员后,预测结果的统计置信度 (P-value) 的分布。该图表明,对超过 50% 的蛋白质可获得 P-value < 0.05 的预测结果。对高置信度区间预测结果的少量抽样实验验证了大多数预测结果是可靠的(龙朋朋等,待发表)。
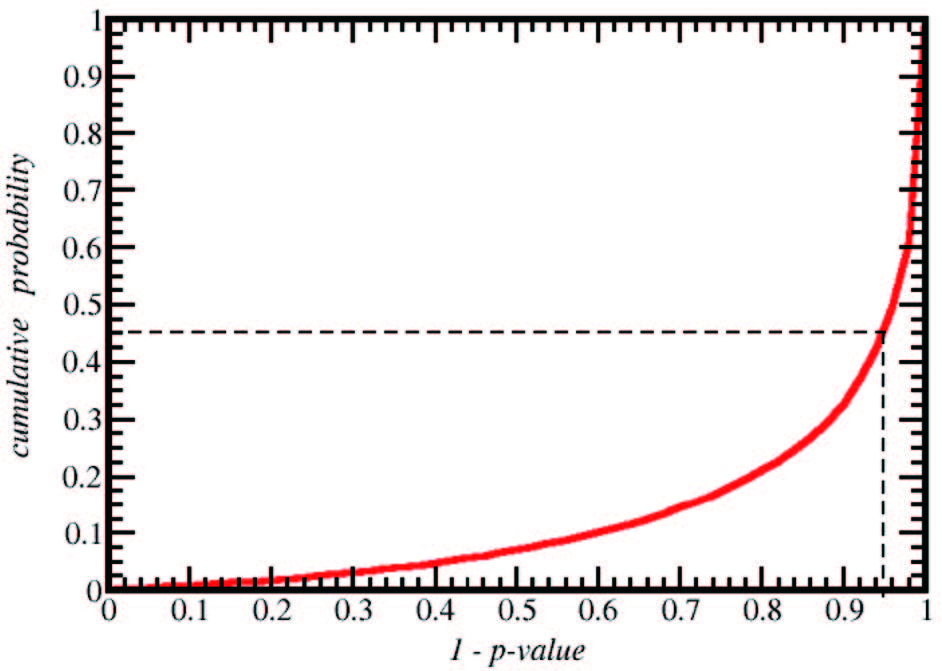
上述基于基因组序列的方法只适用 tetR 家族成员,且各项假设都成立时才能做出有效的预测。文献报道中有一些从原理来看更通用的方法,例如基于蛋白质 -DNA 复合物的结构模型,直接从 DNA 结合结构域的氨基酸序列出发做出预测。这类方法目前的准确性怎样呢,我们考察了 footprintDB web 服务器。该服务器整合了多种根据 DNA 结合结构域的氨基酸序列预测 DNA 结合位点的模型,其中一些模型用实验测定的蛋白质 -DNA结合数据校准过。作为测试,我们从前文基于基因组序列预测可得到高置信度结果的转录因子中选择了数百个,用该服务器预测了其 DNA 结合位点。结果发现,对大部分用于查询的(约 80%)转录因子,footprintDB 给出的预测结果可能是不正确的:预测出的 DNA 序列与前述基于基因组预测的序列无相似性,与已得到实验验证的结果也不一致。这表明,现有的预测转录因子或其他蛋白质的 DNA 识别序列的通用方法,预测效果并不理想。通用性和准确性都较好的计算方法还有待发展。
尽管基于基因组序列的预测方法通用性有限,但它们能对数以千计的天然转录因子给出较为可靠的结果 (图 1)。今后,这些基于数据的结果也有可能用来改善通用的、基于蛋白质序列的预测方法。图 2展示了 tetR 家族 6 个不同成员与 DNA 复合物的晶体结构。尽管这些蛋白质的 DNA结合结构域序列差别大,识别的 DNA 序列多样,但复合物中蛋白质 -DNA 相互作用部分结构是高度保守的。这样,从原理来说, DNA 序列应该由这些结构高度保守的 DNA 结合结构域的氨基酸序列决定。然而,如果用现有的分子力场等关于分子间相互作用的物理模型来进行预测,这类模型还难以准确辨别序列变化引起的亲和力变化;如果要使用机器学习等数据驱动的方法,仅仅依靠少数已知的复合结构和少量与序列变化相关的实验结果也难以构建可靠的定量模型。在今后研究中,如能整合基于基因组序列的预测数据和如图 2 所示的结构数据,采用机器学习等人工智能方法,有可能建立比基于基因组序列的方法更加通用,同时比现有基于蛋白质序列和结构的方法更准确的计算工具。
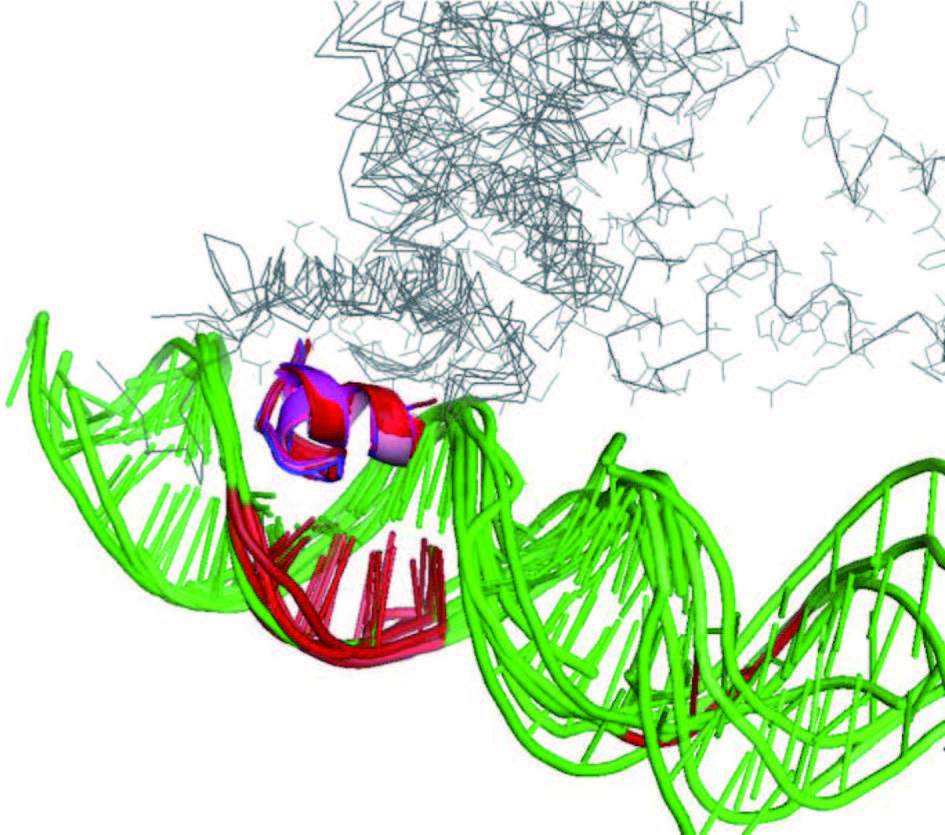
图2 六个 tetR 家族成员蛋白与 DNA 复合物的结构叠合图
2.3 预测小分子结合能力
在接下来的例子中,我们考察预测转录因子、酶等蛋白质元件对特定小分子的结合能力。目标小分子化合物是给定的,我们要从一系列天然蛋白质元件中预测哪些蛋白质有能够识别该小分子的口袋,这被称为反向对接 (inverse docking)问题。
基于现有计算工具,有两条可能技术路线实现反向对接。第一条路线以受体蛋白的结构为中心,将基于结构的分子对接 (molecular docking)算法逐个应用于候选蛋白质元件,预测和评估它与小分子的结合。采用这一路线需要知道每个候选蛋白质元件的空间结构。如果没有实验数据,则需要先使用比较建模等结构预测工具预测其空间结构。多种因素可能影响最终预测结果的准确性,包括:比较建模预测的受体结构是否准确;受体在结合小分子后是否可能发生大的构象变化;分子对接是否能找到最优结合模式;用于评估亲和力的评分函数是否足够准确,等等。第二条技术路线是以小分子为中心。我们可以用化学信息学工具比较目标小分子和其他已知的,能够与不同已知序列或结构的蛋白质相互作用的小分子。这种比较既可以基于小分子间的整体相似性,也可以基于它们的化学子结构的相似性。根据与相同或相似的小分子相互作用的蛋白质 (模板蛋白)与候选蛋白质元件在某些方面的相似或差异性,我们可预测后者识别目标小分子的可能性。如果已知数据足够充分,我们可比较模板蛋白质和候选蛋白质元件在结合口袋周围的空间结构细节,据此做出的预测有可能达到较高的准确性。即使是其他情形,预测结果对后续实验设计 (如优先考虑哪些候选元件进行实验筛选)也可具有指导意义。
在候选蛋白质元件数量不是特别多 (如用其他计算工具筛选过后)的情况下,可以使用分子模拟技术进行更细致的计算筛选。对小分子与每一种候选蛋白质可能形成的复合物,我们构建初始空间结构模型 (用比较建模、分子对接等工具完成)。按分子模拟要求,先对体系进行初始优化、平衡,再通过求解牛顿运动方程,得到体系结构 (原子空间坐标)随时间演化的轨迹。通过对结构和相互作用等特性的时间轨迹进行分析,可判断蛋白质 -小分子复合物的合理性。分析中可考虑的一些主要特征包括:小分子是否稳定结合于预期结合位点;是否有足够的特异性相互作用 (氢键、盐键、疏水相互作用等)维系结合;这些相互作用在模拟过程中是否稳定,等等。分子模拟的计算代价相对较大,在现有的大多数多核计算服务器上,对每个候选蛋白的模拟分析可能花费若干小时或更长的计算时间。随着计算机硬件速度和并行规模的快速提升,这一工具的应用会越来越广泛。
2.4 结合和催化口袋的理性设计
除预测和筛选蛋白质元件外,计算方法也可以用于设计和改造蛋白质元件的小分子结合位点,或酶的活性中心。目前,最广为人知,并且已有一些成功例子的理性设计方法是 RosettaDesign。近期国际上两项研究把设计和实验结合,分别成功改变了两种转录因子的诱导物特异性。前面提到的通过从头设计获得有能被观察到的催化活性的酶的例子也使用了同类方法。这些例子包括分别催化 Kemp 消除反应、逆向醛缩反应和双分子 Diels-Alder 反应的人工酶。这类设计的主要步骤可概括为:确定结合口袋的关键残基 (直接参与催化、与小分子配体发生特异性相互作用等的残基)的构型,得到理论口袋或理论酶;通过几何匹配确定理论口袋中关键残基在蛋白质主链骨架上可能的位置;设计口袋周围其他位置的氨基酸残基。对于酶设计,理论酶的构型可能需要采用量子化学计算来确定。在设计完成后,可以用分子模拟进一步验证结果。近期,基于分子模拟的酶设计工作表明,模拟过程中活性中心氢键网络的完整性和稳定性可作为重要评价指标。
目前,对小分子结合口袋和酶活性中心计算设计的成功率还不高,这与算法中采用的一些假设和近似有关。其一是使用量子化学计算得到的理论口袋 /理论酶不一定对应于复杂蛋白环境中最有利的相互作用构型。例如,通过水分子介导的相互作用难以被处理。在这一点上,用结构生物信息学、化学信息学工具进行数据驱动的设计可以很好地补充计算化学方法。随着蛋白质 -小分子复合物高分辨结构数据越来越多,数据驱动方法也应能更有效地从数据中提取蛋白质 -小分子的相互作用特征,并据此设计更合理的结合口袋。此外,现有算法的一个主要缺陷是使用几何算法把理论获得的活性中心匹配到结构固定的主链骨架上。由于缺乏对主链骨架空间构象变化的描述,该算法只能勉强找到近似的匹配,后续设计的结合口袋或活性中心很难以较理想的方式与配体相互作用。这一点在对设计结果的实验解析结构中往往很明显:预期的氢键等特异性相互作用并未呈现或不处于有利构型。克服这一困难需要考虑完全柔性的主链骨架。
在常规的分子模拟中,蛋白质构象是完全柔性的,但所使用的基于物理模型的分子力场能量函数依赖于侧链原子、溶剂分子等,不适用于侧链处于待定状态的蛋白质设计问题。在结构建模中使用的另一类能量模型是统计能量模型,它来源于对数据库的统计分析。在近期研究中,我们发现在恰当的统计能量模型下,只考虑主链也能建立逼真的蛋白质主链结构。该已发表的模型还只考虑了刚性二级结构片段之间的堆叠,没有考虑构象柔性。更进一步,我们建立了一种能考虑全部构象柔性的新型统计能量模型 (黄斌等,待发表 )。用随机动力学模拟等分子模拟技术可以在该能量函数下对主链骨架进行连续采样。与此同时,我们还发展了一种动态组装方法,可以在对主链骨架构象采样的同时,把理论设计的活性中心所包含的各种氨基酸残基定位到合适的位点上,保持它们与配体之间的相对构型和重要相互作用。图 3 示例了用此方法把丝氨酸水解酶活性中心定位到一个完全柔性的蛋白质骨架上的初步设计结果。理论上,对催化重要的特异性相互作用在设计得到的构型下都被保留了,而主链结构的合理性是由统计能量模型来保证的。需指出的是,图 3 还只是一个示例,在方法和结果的多个方面都尚待优化,其目的是为了说明考虑骨架柔性的活性中心设计可能达到的效果。
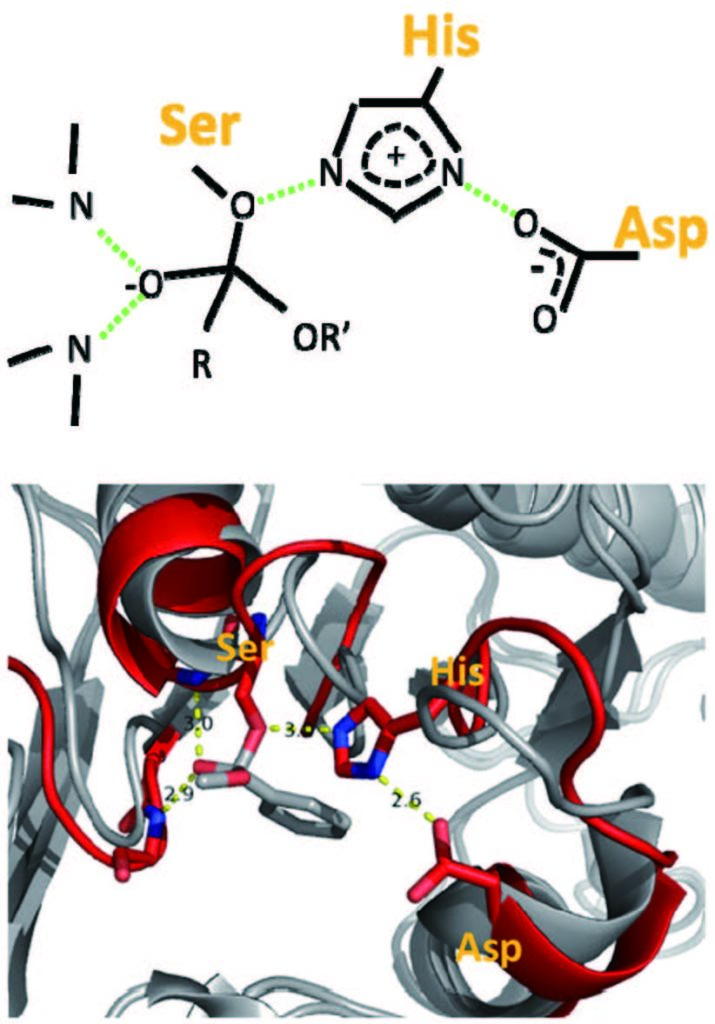
图3 在柔性主链构象采样过程中把丝氨酸水解酶活性中心(上)定位到蛋白质骨架上(下)
3 展望
在蛋白质元件预测和设计中,计算已经展示了其通过整合利用多源、大量数据信息,提供预测结果指导筛选恰当的实验对象以及提供设计结果提高实验效率和成功率等重大作用。尽管如此,目前无论是计算方法的发展还是计算的应用都还远不充分。在建立通用性高的预测方法和提高理性设计的成功率等方面,现有的计算工具还存在一些困难。但是,这些困难正在逐步得到克服,这正是计算的潜力所在。克服这些困难的途径包括:已有的计算工具以更恰当的方式得到应用,例如分子模拟应用于蛋白质设计中;结合更大量、更高质量的数据和恰当的机器学习、统计学习等推动数据驱动方法的发展;数据驱动方法与计算化学方法,如量子化学、分子模拟等的结合,等等。随着更多的研究人员重视和投身于此领域的研究,计算机硬件性能持续提升,越来越多对合成生物学有意义的天然蛋白质元件性质将得到准确的预测;借助合理设计的元件,一些仅用天然元件无法实现的合成生物系统将得以实现。