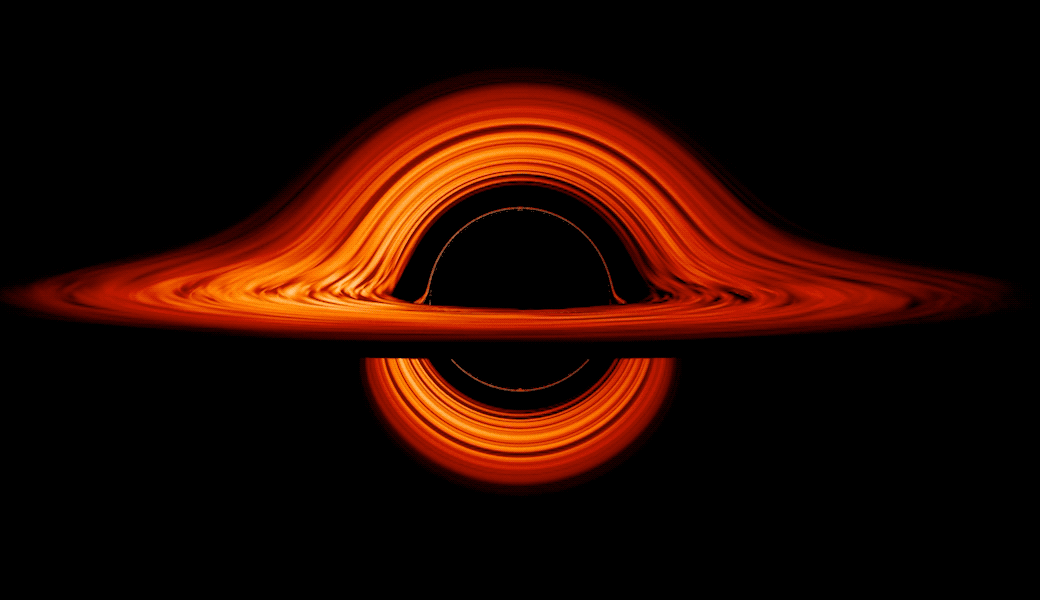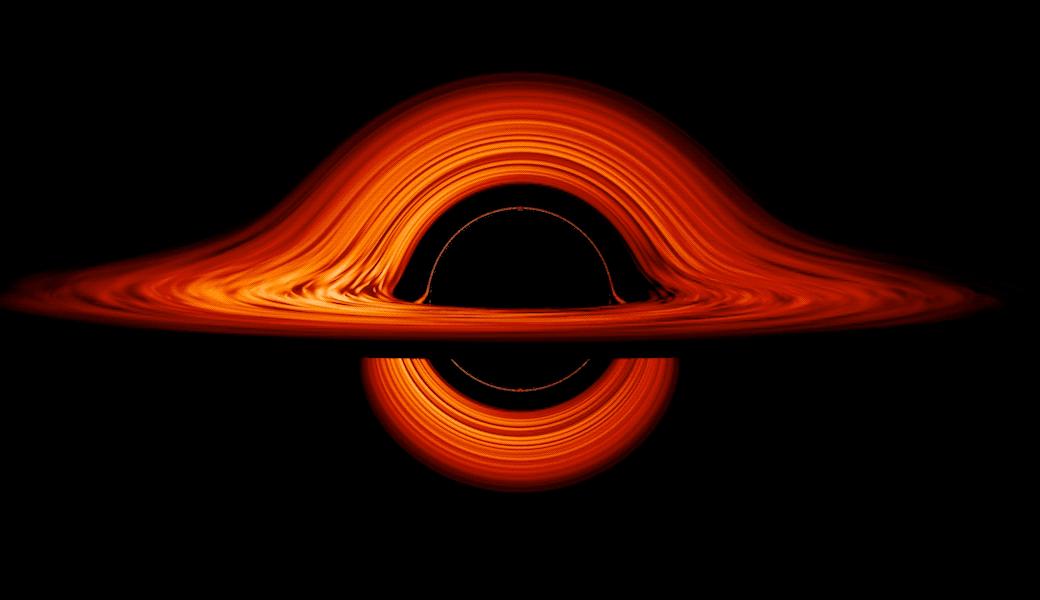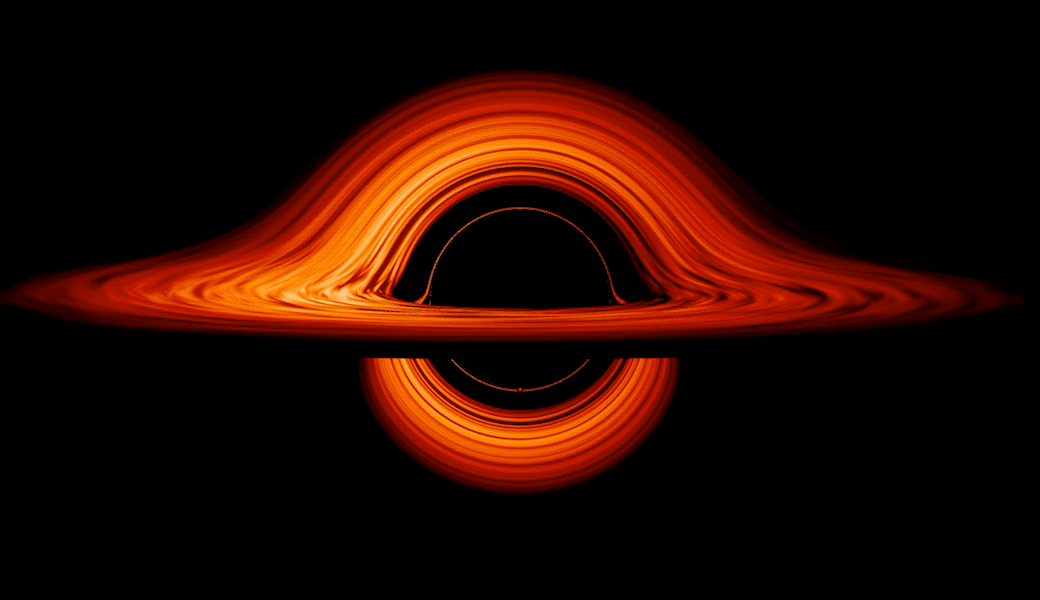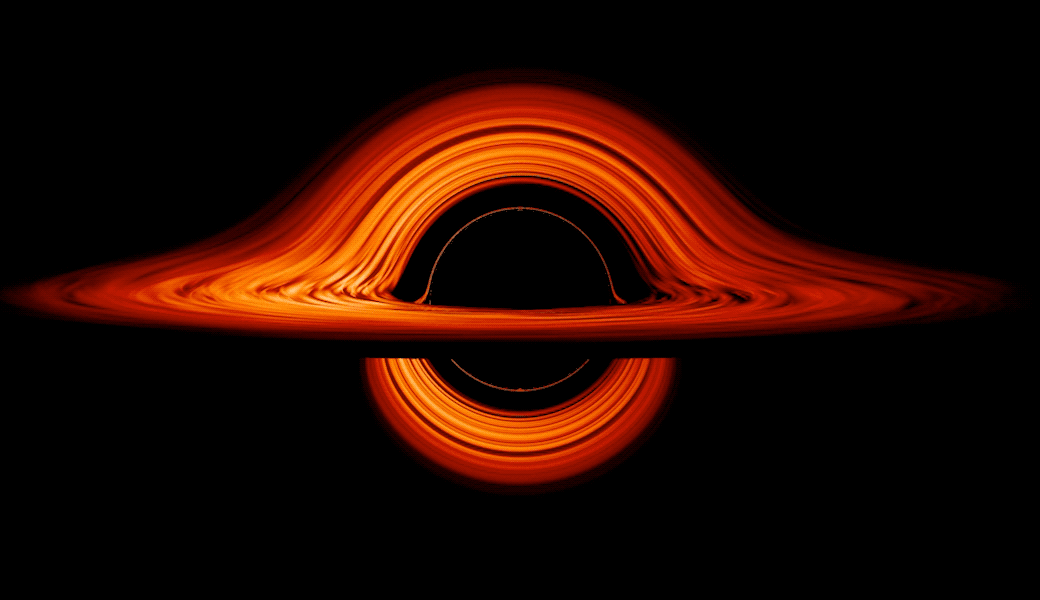
This new visualization of a black hole illustrates how its gravity distorts our view, warping its surroundings as if seen in a carnival mirror. The visualization simulates the appearance of a black hole where infalling matter has collected into a thin, hot structure called an accretion disk. The black hole’s extreme gravity skews light emitted by different regions of the disk, producing the misshapen appearance.
Bright knots constantly form and dissipate in the disk as magnetic fields wind and twist through the churning gas. Nearest the black hole, the gas orbits at close to the speed of light, while the outer portions spin a bit more slowly. This difference stretches and shears the bright knots, producing light and dark lanes in the disk.
Seen nearly edgewise, the turbulent disk of gas churning around a black hole takes on a crazy double-humped appearance. The black hole’s extreme gravity alters the paths of light coming from different parts of the disk, producing the warped image. The black hole’s extreme gravitational field redirects and distorts light coming from different parts of the disk, but exactly what we see depends on our viewing angle. The greatest distortion occurs when viewing the system nearly edgewise.
Viewed from the side, the disk looks brighter on the left than it does on the right. Glowing gas on the left side of the disk moves toward us so fast that the effects of Einstein’s relativity give it a boost in brightness; the opposite happens on the right side, where gas moving away us becomes slightly dimmer. This asymmetry disappears when we see the disk exactly face on because, from that perspective, none of the material is moving along our line of sight.
Closest to the black hole, the gravitational light-bending becomes so excessive that we can see the underside of the disk as a bright ring of light seemingly outlining the black hole. This so-called “photon ring” is composed of multiple rings, which grow progressively fainter and thinner, from light that has circled the black hole two, three, or even more times before escaping to reach our eyes. Because the black hole modeled in this visualization is spherical, the photon ring looks nearly circular and identical from any viewing angle. Inside the photon ring is the black hole’s shadow, an area roughly twice the size of the event horizon — its point of no return.
“Simulations and movies like these really help us visualize what Einstein meant when he said that gravity warps the fabric of space and time,” explains Jeremy Schnittman, who generated these gorgeous images using custom software at NASA’s Goddard Space Flight Center in Greenbelt, Maryland. “Until very recently, these visualizations were limited to our imagination and computer programs. I never thought that it would be possible to see a real black hole.” Yet on April 10, the Event Horizon Telescope team released the first-ever image of a black hole’s shadow using radio observations of the heart of the galaxy M87.
克服上述困难有两条可能的途径。第一条途径是,从自然界中存在的大量具有不同功能,特别是具有不同特异性分子识别能力的天然蛋白质中找出适当的元件。例如,同一家族的不同转录因子可响应不同的化学信号,也可识别不同的操纵子序列;催化同类化学反应的酶可能有不同的底物专一性和反应特异性,等等。另一条途径是,采用蛋白质设计和定向进化等手段,改变天然蛋白质分子的功能活性,甚至重新设计蛋白质,获得适用的元件。例如,通过诱导物结合位点或 DNA 结合位点改造,转录因子可以响应新的化学诱导信号或识别新的 DNA 结合位点;酶的底物特异性可通过蛋白质工程改变等。
除发现有所需功能的蛋白质元件外,蛋白质元件的改造,乃至重新设计也可以极大受益于计算。对天然蛋白质进行改造的目标包括调控稳定性、改变环境偏好性、改变相互作用特异性 (如转录因子识别新的诱导物分子或 DNA 序列、酶催化新的底物等 )等;在一些应用中,可能还必须获得自然界不存在的全新功能的蛋白质,如催化新反应类型的酶。从发展的角度看,解决后一类问题是合成生物学走出天然体系的局限,达到“超越自然”目标的重要途径。
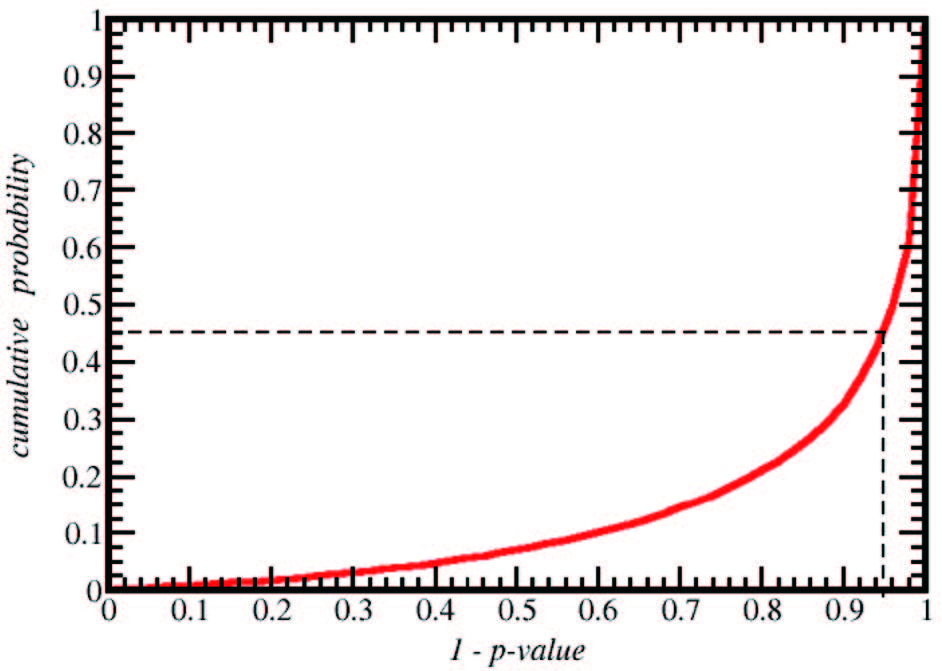
首先,我们考察一类基于基因组序列预测转录因子与 DNA 结合序列的方法。该方法基于以下现象或假设:被 tetR 家族蛋白识别的 DNA 序列具有回文特征;蛋白质识别的 DNA 序列在基因组上频繁出现在相应蛋白质基因位点附近;蛋白质 DNA 结合结构域的氨基酸序列决定识别位点半回文区的 DNA 序列。基于这些假设,可相对容易地预测一些 tetR 成员的 DNA 识别位点。我们把这个过程流程化、程序化,建立计算工具,从而能对大量 tetR 家族成员做出自动预测(龙朋朋等,待发表)。这里,简单把手工流程翻译成程序不足以实现高鲁棒性的自动计算。流程自动化过程中需要考虑的问题包括:每个转录因子基因位点附近都可以找到大量的回文序列片段,但其中绝大部分 (或全部 )都不是我们要寻找的位点;要确定真正的结合位点,必须考虑其他含同源蛋白的基因组中回文序列片段在目标区域被富集的情况,在此过程中,我们需要排除目标蛋白编码区以外基因组同源性的影响;此外,回文序列的判别会受到序列碱基组成的影响,如 G、C 含量高的片段容易被识别为回文序列,且在基因组中出现频率高,等等。综合处理好这些因素后,程序化的流程能产生可重复、可靠的结果,而无需依赖手工筛选 (手工筛选难以做到高通量预测 )。图 1显示了把这一自动化流程应用于基因组序列已知,可从公共蛋白数据库中找到的全部 tetR 家族成员后,预测结果的统计置信度 (P-value) 的分布。该图表明,对超过 50% 的蛋白质可获得 P-value < 0.05 的预测结果。对高置信度区间预测结果的少量抽样实验验证了大多数预测结果是可靠的(龙朋朋等,待发表)。
图1 基于基因组序列预测 tetR 家族成员蛋白的 DNA 识别序列得到的最大预测分数的 p-value 分布
上述基于基因组序列的方法只适用 tetR 家族成员,且各项假设都成立时才能做出有效的预测。文献报道中有一些从原理来看更通用的方法,例如基于蛋白质 -DNA 复合物的结构模型,直接从 DNA 结合结构域的氨基酸序列出发做出预测。这类方法目前的准确性怎样呢,我们考察了 footprintDB web 服务器。该服务器整合了多种根据 DNA 结合结构域的氨基酸序列预测 DNA 结合位点的模型,其中一些模型用实验测定的蛋白质 -DNA结合数据校准过。作为测试,我们从前文基于基因组序列预测可得到高置信度结果的转录因子中选择了数百个,用该服务器预测了其 DNA 结合位点。结果发现,对大部分用于查询的(约 80%)转录因子,footprintDB 给出的预测结果可能是不正确的:预测出的 DNA 序列与前述基于基因组预测的序列无相似性,与已得到实验验证的结果也不一致。这表明,现有的预测转录因子或其他蛋白质的 DNA 识别序列的通用方法,预测效果并不理想。通用性和准确性都较好的计算方法还有待发展。
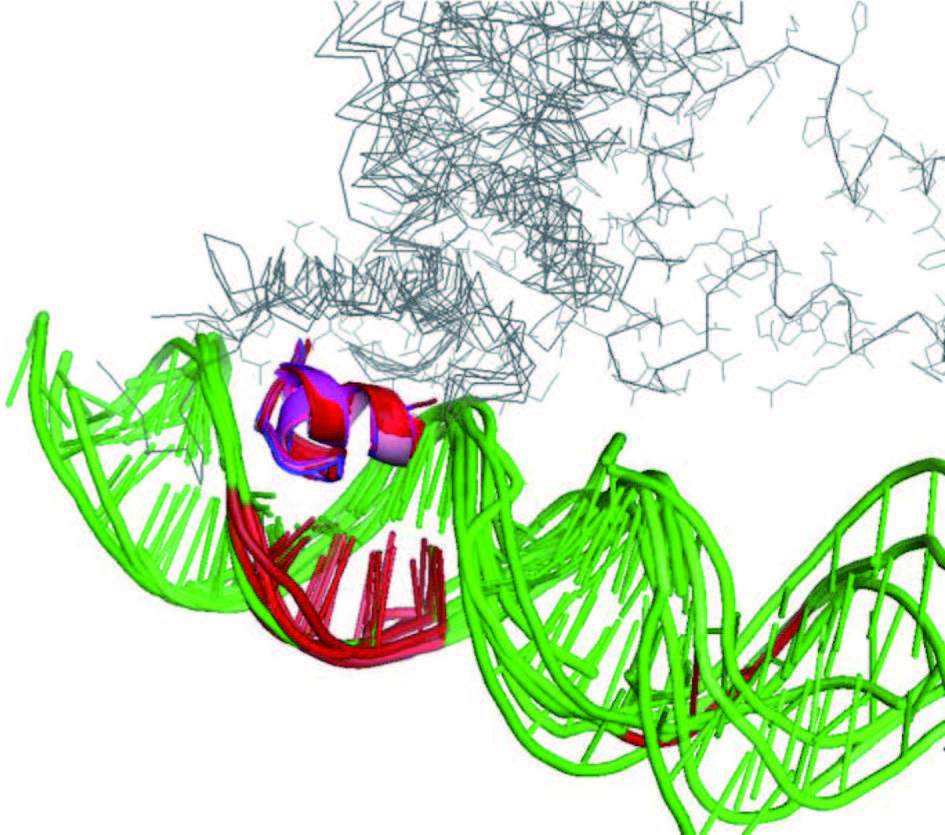
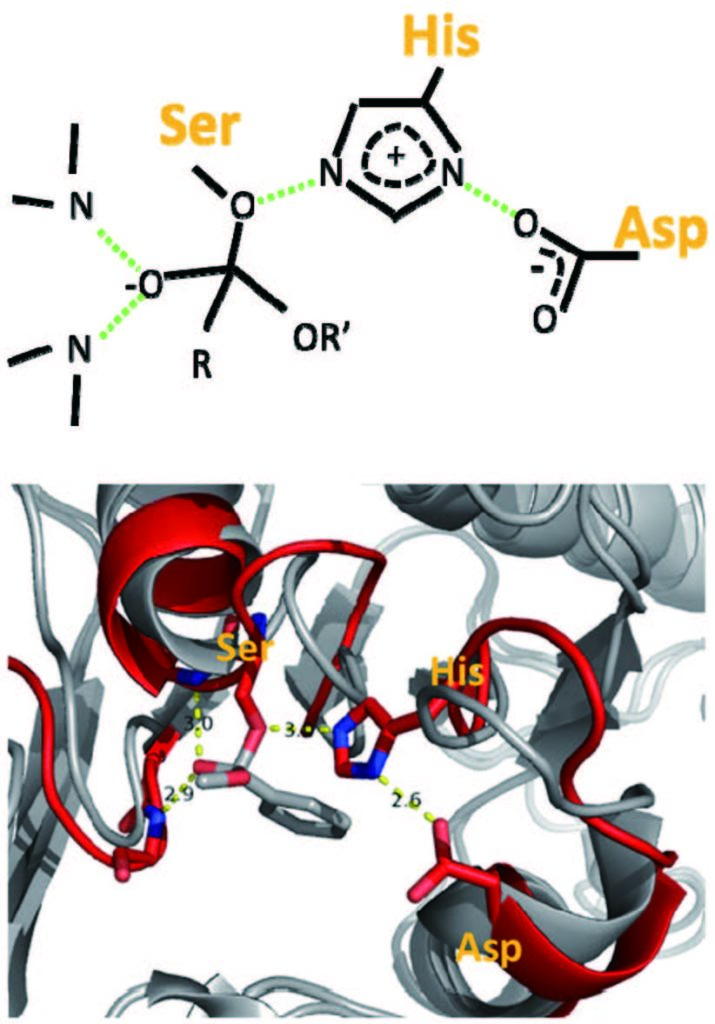
尽管基于基因组序列的预测方法通用性有限,但它们能对数以千计的天然转录因子给出较为可靠的结果 (图 1)。今后,这些基于数据的结果也有可能用来改善通用的、基于蛋白质序列的预测方法。图 2展示了 tetR 家族 6 个不同成员与 DNA 复合物的晶体结构。尽管这些蛋白质的 DNA结合结构域序列差别大,识别的 DNA 序列多样,但复合物中蛋白质 -DNA 相互作用部分结构是高度保守的。这样,从原理来说, DNA 序列应该由这些结构高度保守的 DNA 结合结构域的氨基酸序列决定。然而,如果用现有的分子力场等关于分子间相互作用的物理模型来进行预测,这类模型还难以准确辨别序列变化引起的亲和力变化;如果要使用机器学习等数据驱动的方法,仅仅依靠少数已知的复合结构和少量与序列变化相关的实验结果也难以构建可靠的定量模型。在今后研究中,如能整合基于基因组序列的预测数据和如图 2 所示的结构数据,采用机器学习等人工智能方法,有可能建立比基于基因组序列的方法更加通用,同时比现有基于蛋白质序列和结构的方法更准确的计算工具。
红色和紫色显示结构高度保守的 DNA 识别 motif 图2 六个 tetR 家族成员蛋白与 DNA 复合物的结构叠合图