
台湾国立中央大学 认知神经科学研究所 陈永仪
月度归档: 2019 年 7 月
三维基因组测序技术发展
内蒙古大学生命科学学院
杨琬婷
北京大学生命科学学院
杨磊 王世强
1 三维基因组
1965 年,Flemming 在细胞分裂之前观察到了染色体的形成。如 20 世纪初 Boveri-Sutton 遗传染色体理论所阐明的那样,染色体在有丝分裂和减数分裂过程中的行为证明了它们在遗传信息携带方面有着重要的作用。多年来,生物学家专注于研究染色体的结构、动力学和行为,希望发现基因表达、转录调控机制的全过程,除了主要基因序列以外,目前已经致力于研究基因组的特征。科学家们已经采用生物化学分析和计算工具来绘制转录活性、转录因子结合位点和对组蛋白的化学修饰以及 DNA本身的位点,最终发现数以万计的转录单位和数百万潜在的人类基因组顺式调控元件 。
然而,转录调控过程不单单是在一维线性核苷酸链上就能进行的。在体内,组蛋白包裹着核苷酸链,组成染色体,形成高度致密的结构。转录调控取决于调控元件之间的物理相互作用,以增强子和启动子为例,在许多基因座位上发现了调节增强子 -启动子环 。在线性意义上它们通常不相邻,但在转录调控中的作用却有非线性相互作用,从两个角度可以阐明这个现象。首先,增强子可以指导线性距离较远的靶基因的表达 ;其次,受增强子影响最大的基因往往并不是线性距离最接近的基因 。随着研究的深入,越来越多的证据表明,这种表面上的“长程”调控是可能的。转录时,尽管 DNA双链中会插入很长的核苷酸序列,增强子也与其靶基因的启动子在物理上接近 。这种物理上的接近,使得增强子与启动子上结合的蛋白质复合物相互作用,从而影响靶基因的转录。
对三维基因组的研究,可以不仅仅局限于功能元件的单一线性作用,而是提高对其在染色质物理结构上相互作用的认识,为了更好地了解染色体相互作用技术,研究者们对许多研究技术进行了革新。
2 三维基因组研究技术
三维基因组研究的初期是建立在显微镜研究上的,如 FISH 和一些其他显微镜方法,这些方法能够进行基因定位的单细胞分析 。然而,在基因组和细胞群体规模上,它们的通量和分辨率有限,因此可能无法高效且清晰地发现核组织的一般原则或个别基因的特征。染色质构象捕获技术在一定程度上打开了染色体相互作用的大门,该技术最初设计用于测量两个基因组座位在三维 (3D) 核空间内相互作用的频率。随着科学的进步和技术的革新,这种方法已经从定性和定量两方面发展,并从检测两个特定基因座的配对相互作用到全基因组染色质相互作用组,再结合对表观遗传染色质背景的分析,揭示了通用基因组折叠原理。
2.1 染色质构象捕获技术及其衍生技术
2002 年,Dekker 等开创性地发展了染色质构象捕获技术(chromatin conformation capture technology, 3C) 以分析染色质的物理特性,应用于研究酵母染色体的折叠。至此,三维基因组的研究进入到了一个新的研究水平,由于需要更高的分辨率与通量,染色质构象捕获技术 (3C) 不断发展,从“一对一” (3C) 衍生为“一对多”(4C) 、“多对多”(5C) ,再到全基因组 (Hi-C 、Micro-C)、靶向区域分析 (ChIA-PET 、Capture-C 、Capture Hi-C ) 等等,这些方法有很大的潜力,也有一定的局限性。
2.1.1 染色质构象捕获技术(chromatin conformation capture technology, 3C)
3C 及 3C 衍生技术的第一步是建立 DNA 的3D 结构的表达模板。完整的细胞核被分离出来后,常常使用固定剂甲醛固定,使 DNA 与蛋白质以及蛋白质与蛋白质之间交联。接下来,用识别 6 个碱基对(bp) 的限制性内切酶,如 HindIII、BglII、SacI、BamHI 或 EcoRI,或识别 4 个碱基对的限制性内切酶,如 AciI 或 DpnII 消化。随后,交联的 DNA 片段的黏性末端在稀释的条件下被重新连接 ( 在交联的片段之间 ),在空间中共定位,线性距离较远的DNA 片段可以以这种方式彼此连接,由此创建模板,其实质可被看作是 3D 核结构的一维 (1D) 投射。最后,确定不同位点已经交联的相对频率,通过对全基因组相互作用频率的分析,获得关于一般核结构的信息以及染色体的物理性质和构象。
其中,在重新连接时,根据实验目的不同,可使用半定量 或定量 PCR 扩增有相互作用的片段,将引物设计在所有可能的限制性片段的末端附近,通过比较不同引物组合的扩增效率,建立连接频率矩阵,来充当成对相互作用频率的标志。
2.1.2 环状染色质构象捕获技术(circular chromatin conformation capture technology, 4C)
4C 最初将 3C 技术与微阵列相结合,分析选定基因组位点与阵列上所有基因组片段的接触 ,该技术可在全基因组范围搜索与核空间中给定基因座接触的 DNA 基因座,其中,活动和非活动基因参与许多远程染色体内相互作用,也可以形成染色体间接触。在 4C 技术中,3C 分析如常进行,但 PCR步骤省略,用第二种限制酶切割并重新环化,然后,通过反向PCR 特异性扩增与该染色体位点接触的所有序列。最后,通过微阵列分析或下一代测序(next generation sequencing, NGS) 方法分析。
2.1.3 染色质构象捕获碳拷贝技术(chromatin conforma- tion capture carbon copy technology, 5C)
在染色体构象捕获碳拷贝 (5C) 中,允许同时测定多个序列之间的相互作用 ,通过常规 3C 产生3C 文库,使用高度多重连接介导的扩增(LMA) ,在多重 PCR 反应中退火并连接 5C 寡核苷酸而转化为 5C 文库。这些连接点的读数可以在微阵列上进行,也可以通过高通量测序进行。5C 已被用于研究 β- 球蛋白基因座 和 α- 球蛋白基因座的染色质构象等。
总得来说,3C 方法本质上依赖于定量测定而非定性测定,其创新之处在于首次提出了这种“邻近连接”思路想法。但是,如果距离超过几千个碱基位点时,特异性连接产物变得非常罕见,无法通过 3C PCR 进行准确定量。4C 是评估个体基因组位点 DNA 接触特征的方法,显示了活性和非活性染色质之间的分离,但仅限于描述与同一条染色体上其他地方 ( 顺式 ) 或其他染色体 ( 反式 ) 的较大区域的长距离接触,相距 50 kb 以上的基因间和基因与功能元件间的相互作用尚不容易被发现。而 5C技术相对于 4C 技术而言,可在有其他接触的情况下,研究特定位点之间的 DNA 接触,该技术广泛适用于大规模基因组元件顺式和反式互作网络的研究以及高阶染色体结构的研究,它不仅可以识别特定位点之间的相互作用,还可以建立整个基因组区域的接触频率矩阵。
2.2 高通量染色质构象捕获技术(high-throughput chromatin conformation capture, Hi-C)
2.2.1 Hi-C
Hi-C 是 3C 的一个高通量版本,用于检测所有基因组位点之间的所有相互作用 ,从而呈现全基因组接触图。在 Hi-C 中,创建 3C 模板的步骤稍作调整。在连接之前,限制性末端用生物素标记的核苷酸填充。在末端连接之后,将 DNA 纯化并剪切,并用亲和素富集被标记的连接接头 ( 双端 ) 进一步分析。最后,可得到整个基因组的片段之间的成对相互作用频率矩阵,其分辨率取决于限制性位点密度和测序深度。
Hi-C 没有特异性,更加细节、罕见的现象常常只能在详细的接触图中找到,这些接触图需要对 Hi-C 文库进行非常深入的测序。除此之外,如果仅集中于特定的基因组位点、特定的基因座,甚至特定类别的序列 ( 如基因启动子、增强子、边界等 ),绝大多数 Hi-C 读数是多余的,因此,Hi-C 文库的测序变得过于昂贵。虽然 Hi-C 能够检测全基因组范围内的长程相互作用,但其有效分辨率 ( 取决于限制性片段和实验灵敏度 ) 阻止了特定相互作用的发现。
2.2.2 原位Hi-C
最初,原位 Hi-C 将 Hi-C 方案与核连接分析 相结合,DNA 使用限制酶消化,在填充 5′-突出端的同时掺入生物素化的核苷酸,定量连接平末端片段,剪切 DNA, 用 Streptavidin Beads 捕获生物素化的连接接头,并用配对末端测序分析得到片段。与 Hi-C 相比,原位连接降低了稀释溶液中随机连接引起的假接触的频率 ;其次,所需时间更短 ;第三,它被识别 4 个碱基对的酶切割,实现更高分辨率和更有效的染色质 DNA 切割。
2.2.3 HiChIP
HiChIP 是一种以蛋白质为中心的染色质构象分析方法,它利用了原位 Hi-C 和转座酶介导的珠子库构建原理。在 HiChIP 中,染色质裂解前,先在细胞核原位建立了长程的 DNA 接触,最大限度地减少可能的假阳性相互作用 ,并大大提高DNA 接触捕获效率。然后,进行 ChIP 来直接捕获与蛋白质相关的长程相互作用。配对末端测序后,可分析鉴定出基因组中两个相距较远的片段是否具有相互作用。
2.2.4 BL-Hi-C (Bridge Linker-Hi-C)
BL-Hi-C 是一种功能强大且广泛适用于分析3D 染色质相互作用的方法。近端染色质和结合因子原位交联,使用限制酶 HaeIII ( 识别“GGCC”的 4 碱基切割酶 ) 消化,HaeIII 比以前使用的其他酶离靶向活性区域更近,配合两步连接、测序等,可以完成不依赖抗体或探针等为前提的富集。BL- Hi-C 切割时使用的 HaeIII 富含 CG 碱基,识别序列长度为 4 bp,可以消化较短的限制性片段,增加特异性蛋白质复合物结合的片段的重新连接概率。因此,BL-Hi-C 高效且具有极高的灵敏度,可用于鉴定涉及调节事件的相对动态的染色质结构。此外,改用 20-bp 生物素标记的接头代替之前使用的生物素 -14-dCTP,可将成本降低到原来的三分之一。
2.2.5 OCEAN-C (open chromatin enrichment and network Hi-C)
OCEAN-C 不依赖抗体和探针,通过整合 FAIRE-seq 和 Hi-C 技术,OCEAN-C 可检测活性顺式调节元件富集的开放染色质的相互作用,用于绘制全局开放染色质相互作用。在添加生物素化的残基和 Hi-C 超声处理之后,苯酚 – 氯仿提取去除核小体的染色质 ( 开放染色质 ),从而特异性富集携带生物素信息的 DNA 片段,然后进行文库构建和高通量测序。
OCEAN-C 是研究开放染色质相互作用及其与基因调控关系的新工具。在 3 种 B 细胞相关细胞系(U266、RPMI8226 和 GM12878) 中使用 OCEAN-C技术,发现 3 种细胞中均有近 1 万个开放染色质相互作用中心 (HOCI),且这些 HOCI 也与超级增强子 (super-enhancer)、H3K4me3 区域有重合,可作为连接点,形成全基因组相互作用的网络。
2.2.6 DLO Hi-C
上述这些 Hi-C 衍生方法提高了染色体构象捕获技术的性能,但仍需要提高信噪比,简化实验步骤,并减少实验和测序成本。华中农业大学曹罡教授团队最近新开发了一种基于简单的酶消化和连接步骤的方法 DLO Hi-C ,该方法使用特异性接头,通过不同的接头组合,配合酶切,得到所需研究的片段,这代替了常规方法中生物素标记和沉淀的步骤,有效地提高了信噪比。
由于原位 Hi-C 是高质量染色体构象捕获的黄金标准,该课题组结合了 DLO Hi-C 和原位 Hi-C 的优势,将切割限制酶改为 MseI,该酶在人类基因组中的靶向位点数目远远超过 MboI 和 HindIII,对于复杂的基因组来说,应该能够捕获更多的染色体接触信息。相比于Hi-C 及其衍生技术,DLO Hi-C 简单、经济实惠、高信噪比且速度快,同时引入了一个早期的质量控制步骤,在测序前可快速评估随机连接噪声的比率,可用于全基因组染色体构象捕获,促进了基因组组装、染色体易位分析和宏基因组学研究。
除了本文提到的技术外,近些年开发了 Hi-C方法的许多变体 :TCC ,通过实施固相连接,TCC 大大提高了信噪比,因此,可以对染色体间相互作用进行详细分析 ;micro-C ,使用微球菌核酸酶代替限制性酶来消化染色质,从而得到核小体层次分辨率的染色体互作图谱 ;RTCC ,TCC 与Tn5 转座酶结合;DNase Hi-C 和靶向DNase Hi-C , DNase I 代替限制性内切酶用于交联 DNA 的断裂;Capture Hi-C ,将 Hi-C 与目标富集相结合; snHi-C , 可检测基因组中染色质特征, 包括Loop、TAD和 compartment 等,且可量化得到单细胞数据。图 1 为一些 3C 及其衍生技术流程图 。
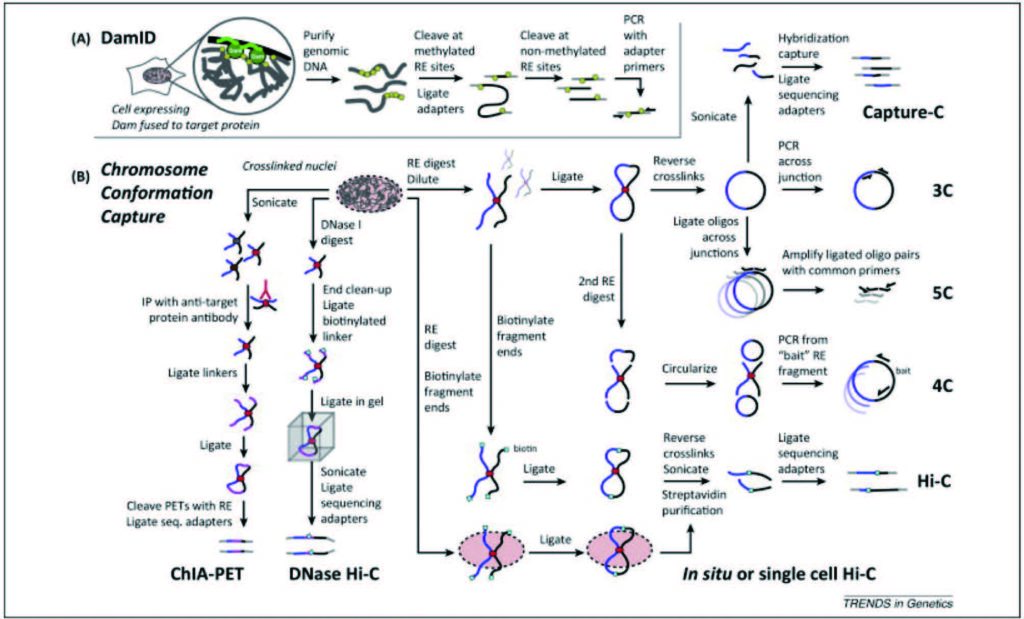
2.3 配对末端标签测序分析染色质相互作用(chromatin interaction analysis using paired end tag sequencing, ChIA-PET)
研究者开发了多种技术用于研究转录因子与转录调控的结合,如染色质免疫沉淀 (ChIP) 芯片 (ChIP-chip) 、ChIP-PET 和 ChIP-Seq 等 ;但这些技术无法确定远端 TF 结合位点的靶基因。3C及其衍生技术更大程度上关注的是 DNA-DNA 接触,虽然它们可以和 ChIP 结合来研究调节基因组三级结构的蛋白质因子结合位点,但无法直接确定结合位点间的相互作用。
配对末端标记测序 (ChIA-PET) 是 3C 技术与 ChIP (ChIP-seq) 的组合。像 ChIP-Seq 实验一样,使用甲醛交联细胞核中的 DNA- 蛋白质复合物,随后通过超声处理将复合物破碎成碎片 ;将所需的与蛋白质结合的 DNA 片段用 ChIP 富集,将富含ChIP 的染色质复合物中的 DNA 片段以两个等分试样与两种不同的半连接寡核苷酸连接 ;将两个等分试样混合,并将邻近半连接物彼此连接 ;反向交联后,复合物中的蛋白质被消化并提取 DNA 片段 ;然后用限制性酶 MmeI 消化去除蛋白质,DNA 片段以“标签 – 接头- 标签”顺序形成配对末端标签 (PET)构建体 ;最后,采用新一代测序设备对 PET 测序,如 Illumina Hi-Seq2500。
基于 Tn5 转座酶可将核邻近连接产物随机片段化并插入到 DNA 中 ,将这两种方法整合在一个反应中,提高了 ChIA-PET 文库构建的整体效率。这种较长的标签长度提高了序列比对的准确性,更重要的是,增加了杂合相定位 SNP 的覆盖率,从而使得染色质相互作用的单体型特异性定位成为可能。
配对末端标记测序 (ChIA-PET) 是一种无偏倚、全基因组、高通量的从头开始的方法。在功能研究中,与 Hi-C 相比,ChIA-PET 与更高分辨率的相关蛋白质关联性更好,且由于使用超声打断 DNA- 蛋白质复合物,产生的噪音较小。
2.4 Capture-C
3C 及其衍生技术有一个主要限制是需要大量的细胞,然而,想要从许多主要组织和稀有细胞群体中取得如此庞大的细胞数目较为困难,因此衍生出了一种低需求技术 Capture-C ,它可以利用少量细胞生成高质量的相互作用图谱。
Capture-C 包含可能发生重大数据丢失的两个关键阶段 :3C 库的准备以及将该材料加工成测序文库,这两个阶段的效率决定了所需的最少细胞数量。Oudelaar 等独立地对两个阶段进行了优化 :优化 3C 文库制备,防止消化和染色质连接过程中的 DNA 损失,以及使用锁相凝胶技术 (phase lock gel technology) 进行DNA 回收;随后访问3C 文库时,开发了两种新的基于 Capture-C 的方法,即低输入 (LI) Capture-C 和 Tag-Capture-C。这些方法使用不同的测序文库制备方案,更适合于减少输入。
2.5 DamID
与传统 3C 技术不同,DamID 没有基于邻近连接,而是采用邻近甲基化的方法,通过将大肠杆菌DNA 腺嘌呤甲基转移酶 (Dam) 连接到染色质蛋白上,Dam 可以与体内靶向蛋白的天然结合位点结合,导致局部 DNA 甲基化,随后可以使用甲基化特异性限制酶或抗体来定位甲基化位点。甲基化的限制性位点相邻,扩增时选择密集的甲基化区域,从而提供与核周边相关的染色质图谱 。
DamID 具有一定的优点。首先,DamID 可以鉴定与特定蛋白质在体内相互作用的序列,可用于检测蛋白质 – 靶标相互作用中的定量差异。使用固定剂交联会引起研究中染色质结构的改变,但腺嘌呤甲基化对 DNA 拓扑结构只有微小的影响 ,并且不会干扰内源蛋白或其靶标的功能。其次,DamID可用于细胞培养和整个生物体。第三,DamID 可以与一些技术结合,如与 PCR 结合进行定量分析,可较为灵敏地检测蛋白质 -DNA 相互作用 ;与DNA 微阵列技术结合使用可在全基因组范围内鉴定特定蛋白质的靶基因。
以上提到以及未详细涉及的方法各有优劣及侧重,是从不同层面、角度揭示染色质的空间结构,解决重要的生物学问题。下表为各项技术的简单对比,应结合实际目的,选择合适的方法。
| 技术 | 通量 | 特点 |
| 3C | 一对一 | 邻近连接 |
| 4C | 一对多 | 反向PCR及高通量测序 |
| 5C | 多对多 | 多个序列的相互作用 |
| Capture-C | 多对所有 | 低样本量 |
| In situ Hi-C | 所有对所有 | 原位连接 |
| Singl cell Hi-C | 所有对所有 | 结合显微操作 |
| BL-Hi-C | 所有对所有 | 不依赖探针与抗体 |
| OCEAN-C | 所有对所有 | 不依赖探针与抗体,高效富集 |
| ChIA-PET | 所有对所有 | 3C技术与ChIP结合,可进行噪声评估 |
| DamID | 所有对所有 | 鉴定与特定蛋白质相互作用的序列 |
| DNase Hi-C | 所有对所有 | DNase I代替限制性核酸 |
3 三维基因组研究的应用
染色质相互作用的形成和维持是由一系列RNA 和蛋白质因子介导的复杂过程,包括 CTCF 、黏连蛋白 和介体复合物 等。目前,许多研究结果已经证明染色质相互作用是细胞调节基因转录的机制,因此,通过研究染色质的相互作用,可能会发现基因表达的调控机制,以及一些疾病的致病机理等等。
在基因表达调控机制研究中,(1) 鉴于 Hi-C 数据中染色体内接触的密度显著高于染色体间接触的密度,Hi-C 数据可用于基因组组装 和染色体易位鉴定 。使用 Hi-C 技术研究基因组取得了一个很重要的突破 :研究人员发现基因组被划分为两个不同类别的染色质区域,一个是开放和活跃的基因组区域 (“A”区室 ),另一个是封闭和非活动基因组区域“( B”区室 )。这些区域可以分成更小的区域,被命名为“拓扑关联区域”(TAD) 。(2) 染色质相互作用除转录激活外,还可以调节抑制。CTCF可以在某些情况下通过阻断增强子 – 启动子相互作用起到转录绝缘体的作用。(3) ChIA-PET、DNase I超敏反应数据和外显子数据的组合显示,染色质相互作用位于外显子存在的区域 。(4) Splinter 等 证明了非编码 RNA (ncRNA) 分子 Xist 能够塑造女性细胞中失活的 X 染色体。
在临床基因组学研究中,(1) 使用 4C 技术来探索 3 号染色体内的反复性倒位和易位导致急性髓性白血病 (AML) 的机制 。(2) 通过 ChIA-PET 鉴定需与 RNA 聚合酶 II 结合维持的染色质相互作用,显示了胚胎干细胞、神经干细胞和神经球干 / 祖细胞之间的差异 ,突出了染色质相互作用的发育特异性。(3) 使用 Hi-C 方法可以分析与各种基因座相关的特定染色质相互作用,应用于 14 个结直肠癌风险位点 。所有 14 个位点显示出显著的长范围 (> 10 kb) 的相互作用,包括 rs6983267 SNP 和 MYC以及 CCAT1 之间已知的相互作用。(4) 癌症与异常的选择性剪接相关,通过全基因组研究,已经发现超过 15 万个剪接体不同的癌症 。因此,异常的染色质相互作用可能导致异常的共转录剪接,导致形成疾病相关的剪接变体,这些病理变异是染色质相互作用可能引发疾病状态的另一种潜在机制。(5)染色质相互作用调节的抑制状态可能由 H3K27me3标记,处于与癌症相关的多梳复合阻遏复合物 (PRC)的 EZH2 组分的控制之下。在一些癌症,如 B 细胞淋巴瘤和骨髓疾病中已经发现 PRC2 突变 。(6)整合 3D 基因组结构已成为许多全基因组关联研究(GWAS) 的关键部分。GWAS 利用连锁不平衡现象来鉴定标记,通常单核苷酸多态性 (SNPs) 与相关疾病的特定性状相关,从基因分型芯片 (genotyping chip) 上的“标记”SNP 中鉴定疾病相关基因座和靶基因。从发表的 GWAS 研究来看,一半或更多的疾病相关 SNP 位于非编码区 。例如,与胰岛素抵抗、II 型糖尿病和冠心病相关的一组 SNP 可能与 RNA 聚合酶 II 相关染色质相互作用的 IRS1 的使用相关 。(7) 将染色质相互作用用作生物标志物 ,其主要优点之一是能够针对性地分析少数基因区域的结构,对生物标志物进行个性化处理,可为个性化基因组分析提供前所未有的解决方案,将染色质相互作用的分析与其他分子技术相结合,也可增强或验证从其他研究结果的特异性。
4 总结
3C 及其衍生技术的成功开发极大地促进了染色质高级构象的研究,人、鼠、酵母、拟南芥、果蝇、大肠杆菌、水稻等一系列物种的三维结构也逐步被报道,得到了分辨率更高的染色体相互作用视图。改进的数据分析策略也使人们对基因组结构更加了解,这些技术不仅用于研究物种全局性的高级构象,同时还可用于比较分析多样本三维构象的差异。
3C 方法本身有一些局限性,其定量测定接触频率的含义实质上是测定交联和片段化 DNA 序列之间的连接频率。显然,评估更多的定量连接结点,测定结果就会更准确。目前,许多验证研究已经表明,连接效率可以被用作接触频率的代表,但仅此而已,由于许多实验因素的影响,不能直接转化为绝对的体内接触频率。
但就现阶段来说,Hi-C 系列方法是获取细胞内染色质相互作用位点的较为全面的首选方法,它会进一步为基因组、转录组、表观基因组等提供更多、更详细的数据,并且在解释疾病相关机制中起到重要作用。Hi-C 及其衍生技术的主要优点是接触频率测定不受抗体沉淀效率的影响,且不受限于反式作用因素,是“所有对所有”的技术。而 Capture-C 与 Hi-C 相比,可以从少量细胞中生成高质量的相互作用图谱,但成本不一定会更低 ;新开发的 DLO Hi-C 简化了 Hi-C 的步骤,简单、低成本且高效。ChIA-PET 最大的特点是可将循环分析用于特定的蛋白质介导的环,聚焦 DNA 与蛋白质复合物的交联。DamID 与这些方法都不太相同,可以通过检测活细胞中相互作用的情况提供与 ChIP 相似的信息。
目前,三维基因组研究技术仍在快速发展中,需要研究者随时关注技术的革新,或者随着研究的深入开发新的技术。总之,选对合适的技术是研究的基础。
5 展望
如本文所述,近年来,在不断发展的 3C 及其衍生技术下,染色质结构领域出现了前所未有的发展,在转录调控、疾病机制等多方面的应用也越来越多。
随着研究的深入,有关技术将进一步被完善、创新。为了得到不同组织结构的特性,对基因组个性化的研究会越来越多,因此须不断改进单细胞全基因组分析的技术以及计算工具,伴随着测序技术的发展,高通量成像领域也会有更多的发展空间。
同时,通过整合来自染色质构象方法和成像技术的数据,3D 基因组图谱在解释自然发生与疾病相关遗传变异中越来越重要。我们需要进一步提高染色质相互作用分析在疾病中的应用,更清晰地了解如癌症等疾病发生时,染色质内、染色之间发生了怎样的变化,以及是否能有效利用以染色质构象结构变化为靶点的基因药物,治疗性地调控染色质相互作用。除此之外,就转化医学而言,不断改进和加强基于染色质相互作用的生物标记,促进利用染色质相互作用来了解与非编码突变和多态性相关的靶基因的个性化医学进展。
尽管染色质相互作用的研究还未全面发展,但有一点很清楚,即未来通过探索染色质动力学和基因组结构变异,基因组学将会在各个方面带给人们意想不到的收获。